15. Marker Gene Method Comparison (Supplemental Figure S39)
15_S39_DE_comparison.Rmd
suppressPackageStartupMessages({
library(ggplot2)
library(ggVennDiagram)
library(Seurat)})
library(countsplit)
library(ClusterDE)We begin by defining several functions. First, the clusterDE workflow.
clusterDE_workflow <- function(seurat_obj, cluster1, cluster2, cores) {
set.seed(1)
original_markers <- FindMarkers(seurat_obj,
ident.1 = cluster1,
ident.2 = cluster2,
min.pct = 0,
logfc.threshold = 0)
seurat_obj_sub <- subset(x = seurat_obj, idents = c(cluster1, cluster2))
count_mat <- GetAssayData(object = seurat_obj_sub, slot = "counts")
synthetic_null <- ClusterDE::constructNull(count_mat, nCores = cores, fastVersion = TRUE)
seurat_obj_null <- CreateSeuratObject(counts = synthetic_null)
seurat_obj_null <- NormalizeData(seurat_obj_null)
seurat_obj_null <- FindVariableFeatures(object = seurat_obj_null)
seurat_obj_null <- ScaleData(object = seurat_obj_null)
seurat_obj_null <- RunPCA(object = seurat_obj_null)
seurat_obj_null <- FindNeighbors(object = seurat_obj_null)
resolution <- 0.1
while (length(levels(Idents(seurat_obj_null))) == 1) {
resolution <- resolution + 0.1
seurat_obj_null <- FindClusters(object = seurat_obj_null, resolution = resolution)
print("Num clusters")
print(length(levels(Idents(seurat_obj_null))))
}
null_markers <- FindMarkers(seurat_obj_null,
ident.1 = 0,
ident.2 = 1,
min.pct = 0,
logfc.threshold = 0)
original_pval <- original_markers$p_val
names(original_pval) <- rownames(original_markers)
null_pval <- null_markers$p_val
names(null_pval) <- rownames(null_markers)
res <- ClusterDE::callDE(original_pval, null_pval, nlogTrans = TRUE, FDR = 0.05)
return(res)
}Then we write a function for the countsplit workflow.
countsplit_workflow <- function(seurat_obj) {
set.seed(1)
counts_mat <- GetAssayData(seurat_obj, "RNA")
split <- countsplit(counts_mat)
Xtrain <- split[[1]]
Xtest <- split[[2]]
seurat_obj_train <- Seurat::CreateSeuratObject(counts = Xtrain)
seurat_obj_test <- Seurat::CreateSeuratObject(counts = Xtest)
# process training data
seurat_obj_train <- Seurat::NormalizeData(seurat_obj_train,
verbose = FALSE)
seurat_obj_train <- Seurat::FindVariableFeatures(seurat_obj_train,
selection.method = "vst",
verbose = FALSE)
seurat_obj_train <- Seurat::ScaleData(seurat_obj_train, verbose = FALSE)
seurat_obj_train <- Seurat::RunPCA(seurat_obj_train,
features = Seurat::VariableFeatures(object = seurat_obj_train),
verbose = FALSE)
seurat_obj_train <- Seurat::FindNeighbors(seurat_obj_train,
verbose = FALSE)
seurat_obj_train <- Seurat::FindClusters(seurat_obj_train,
resolution = 0.8,
verbose = FALSE)
seurat_obj_train <- Seurat::RunUMAP(seurat_obj_train, dims=1:10)
seurat_obj_test <- Seurat::NormalizeData(seurat_obj_test,
verbose = FALSE)
seurat_obj_test <- Seurat::FindVariableFeatures(seurat_obj_test,
selection.method = "vst",
verbose = FALSE)
seurat_obj_test <- Seurat::ScaleData(seurat_obj_test, verbose = FALSE)
seurat_obj_test <- Seurat::RunPCA(seurat_obj_test,
features = Seurat::VariableFeatures(object = seurat_obj_test),
verbose = FALSE)
seurat_obj_test <- Seurat::FindNeighbors(seurat_obj_test,
verbose = FALSE)
seurat_obj_test <- Seurat::RunUMAP(seurat_obj_test, dims=1:10)
Idents(seurat_obj_test) <- Idents(seurat_obj_train)
seurat_obj_test@meta.data$cell_ontology_class <- seurat_obj@meta.data$cell_ontology_class
seurat_obj_train@meta.data$cell_ontology_class <- seurat_obj@meta.data$cell_ontology_class
return(list("seurat_obj_train"=seurat_obj_train, "seurat_obj_test"=seurat_obj_test))
}We write function for plotting the UMAPs.
DE_comparisons_scatter_plots <- function(tissue, tissue_name, legend_pos=c(0.2, 0.2)) {
louvain_default <- custom_scatter(tissue, "umap", group_by = "seurat_clusters", x_title = "UMAP 1", y_title = "UMAP 2", pt.size = 2, label=FALSE) #+ Seurat::NoLegend()
louvain_recall <- custom_scatter(tissue, "umap", group_by = "recall_idents", x_title = "UMAP 1", y_title = "UMAP 2", pt.size = 2) #+ Seurat::NoLegend()
louvain_countsplit <- custom_scatter(tissue, "umap", group_by = "countsplit_idents", x_title = "UMAP 1", y_title = "UMAP 2", pt.size = 2) #+ Seurat::NoLegend()
cell_ontology <- custom_scatter(tissue, "umap", group_by = "cell_ontology_class", x_title = "UMAP 1", y_title = "UMAP 2", pt.size = 2) +
ggplot2::theme(legend.position = legend_pos,
legend.text = ggplot2::element_text(size=20)) +
ggplot2::guides(colour = ggplot2::guide_legend(override.aes = list(size=6), ncol = 1)) +
ggplot2::scale_colour_discrete(na.translate = F)
column_label_1 <- patchwork::wrap_elements(panel = grid::textGrob('Cell Ontology', gp = grid::gpar(fontsize = 64)))
column_label_2 <- patchwork::wrap_elements(panel = grid::textGrob('Seurat Default', gp = grid::gpar(fontsize = 64)))
column_label_3 <- patchwork::wrap_elements(panel = grid::textGrob('recall+ZIP', gp = grid::gpar(fontsize = 64, fontfamily = "Courier")))
column_label_4 <- patchwork::wrap_elements(panel = grid::textGrob('countsplit', gp = grid::gpar(fontsize = 64, fontfamily = "Courier")))
umap_grid <- column_label_1 + column_label_2 + column_label_3 + column_label_4 +
cell_ontology + louvain_default + louvain_recall + louvain_countsplit +
patchwork::plot_layout(widths = c(5, 5, 5, 5),
heights = c(1,3))
return(umap_grid)
}We write function for plotting the P-values.
compare_markers_p_vals <- function(default_markers_a, default_markers_b, xlabel, ylabel, title, xmax, ymax) {
# sort genes alphabetically
default_markers_a <- default_markers_a[ order(row.names(default_markers_a)), ]
default_markers_b <- default_markers_b[ order(row.names(default_markers_b)), ]
log_p_val_a <- -log(default_markers_a$p_val)
log_p_val_b <- -log(default_markers_b$p_val)
df <- data.frame(log_p_val_a, log_p_val_b)
r_p_val <- cor(log_p_val_a, log_p_val_b)
large_text_size <- 32
small_text_size <- 24
correlation_text_size <- 16
p_value_scatterplot <- ggplot2::ggplot(df, ggplot2::aes(x = log_p_val_a, y = log_p_val_b)) +
ggplot2::geom_point() +
ggplot2::theme_bw() +
xlim(0, xmax) +
ylim(0, ymax) +
ggplot2::xlab(xlabel) +
ggplot2::ylab(ylabel) +
ggplot2::annotate("text", x = 25, y = ymax * 0.82, label = paste0("r = ", round(r_p_val, 3)), size = correlation_text_size) +
ggplot2::theme(axis.text = ggplot2::element_text(size = small_text_size),
axis.title = ggplot2::element_text(size = large_text_size),
plot.title = ggplot2::element_text(size = large_text_size, hjust = 0.5)) +
ggtitle(title)
return(p_value_scatterplot)
}We write function for plotting the q-values.
compare_markers_q_vals <- function(default_markers_a, default_markers_b, xlabel, ylabel, title, xmax, ymax) {
# convert clusterDE results tibble to dataframe
default_markers_b <- data.frame(default_markers_b)
# make rownames the Genes
rownames(default_markers_b) <- default_markers_b$Gene
# sort genes alphabetically
default_markers_a <- default_markers_a[ order(row.names(default_markers_a)), ]
default_markers_b <- default_markers_b[ order(row.names(default_markers_b)), ]
log_p_val_a <- -log(default_markers_a$q)
log_p_val_b <- -log(default_markers_b$p_val)
df <- data.frame(log_p_val_a, log_p_val_b)
r_p_val <- cor(log_p_val_a, log_p_val_b)
large_text_size <- 32
small_text_size <- 24
correlation_text_size <- 16
p_value_scatterplot <- ggplot2::ggplot(df, ggplot2::aes(x = log_p_val_a, y = log_p_val_b)) +
ggplot2::geom_point() +
ggplot2::theme_bw() +
xlim(0, xmax) +
ylim(0, ymax) +
ggplot2::xlab(xlabel) +
ggplot2::ylab(ylabel) +
ggplot2::annotate("text", x = 2, y = 340, label = paste0("r = ", round(r_p_val, 3)), size = correlation_text_size) +
ggplot2::theme(axis.text = ggplot2::element_text(size = small_text_size),
axis.title = ggplot2::element_text(size = large_text_size),
plot.title = ggplot2::element_text(size = large_text_size, hjust = 0.5)) +
ggtitle(title)
return(p_value_scatterplot)
}We write function for plotting the marker gene Venn diagram.
marker_gene_venn_diagram <- function(recall_marker_genes, countsplit_marker_genes, clusterDE_marker_genes) {
p <- ggVennDiagram(list("recall+ZIP"=recall_marker_genes,
"countsplit"=countsplit_marker_genes,
"clusterDE"=clusterDE_marker_genes),
label = "count") +
scale_fill_gradient(low = "white", high = "grey") +
guides(fill="none") +
scale_x_continuous(expand = expansion(mult = .2))
return(p)
}Now, we load the limb muscle tissue.
limb_muscle <- readRDS("Limb_Musclecluster_results_seurat.rds")
limb_muscle <- Seurat::ScaleData(limb_muscle, features = rownames(limb_muscle))
# make cell type labels a factor
limb_muscle@meta.data$cell_ontology_class <- as.factor(limb_muscle@meta.data$cell_ontology_class)
#sort cluster labels by cluster size
sorted_limb_muscle_clusters <- names(sort(summary(as.factor(na.omit(limb_muscle@meta.data$cell_ontology_class))), decreasing = TRUE))
limb_muscle@meta.data$cell_ontology_class <- factor(limb_muscle@meta.data$cell_ontology_class, levels = sorted_limb_muscle_clusters)Now, we run the countsplit workflow.
countsplit_res <- countsplit_workflow(limb_muscle)
limb_muscle_test <- countsplit_res$seurat_obj_test
limb_muscle@meta.data$countsplit_idents <- Idents(limb_muscle_test)Plot the UMAP of different clusterings used in this figure.
umap_grid <- DE_comparisons_scatter_plots(limb_muscle, "Limb Muscle")
ggsave("DE_marker_comparison_umap_grid.png", umap_grid, width = 36, height = 10, units = "in")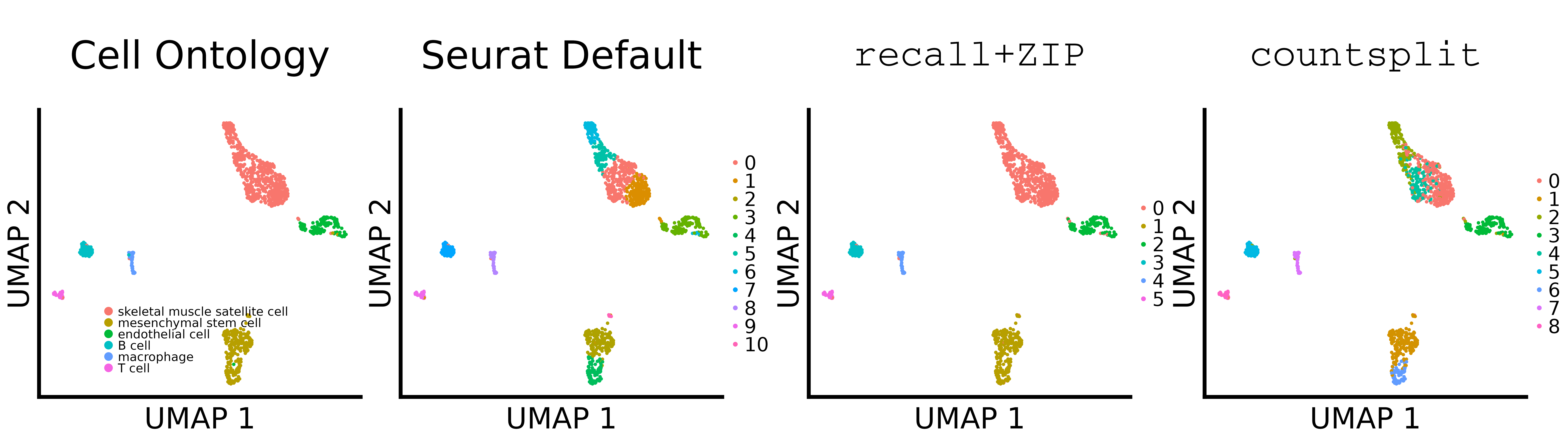
Get marker genes for recall and countsplit
# for countsplit cluster 0 is "skeletal muscle satellite cell" and cluster 1 is "mesenchymal stem cell"
countsplit_markers_0vs1 <- FindMarkers(limb_muscle_test, ident.1 = 0, ident.2 = 1, min.pct = 0, logfc.threshold = 0)
# for recall cluster 0 is "skeletal muscle satellite cell" and cluster 1 is "mesenchymal stem cell"
recall_markers_0vs1 <- FindMarkers(limb_muscle, ident.1 = 0, ident.2 = 1, group.by = "recall_idents", min.pct = 0, logfc.threshold = 0)Run the ClusterDE workflow
# for seurat default cluster 1 is "skeletal muscle satellite cell" and cluster 2 is "mesenchymal stem cell"
clusterDE_res <- clusterDE_workflow(limb_muscle, 1, 2, cores=1)Plot the p-value and q-value scatterplots.
countsplit_vs_recall_markers_plot <- compare_markers_p_vals(countsplit_markers_0vs1,
recall_markers_0vs1,
xlabel = "countsplit -log P-values",
ylabel = "recall+ZIP -log P-values",
title = "Skeletal muscle satellite cell cluster vs\nMesenchymal stem cell cluster",
xmax = 200,
ymax = 400)
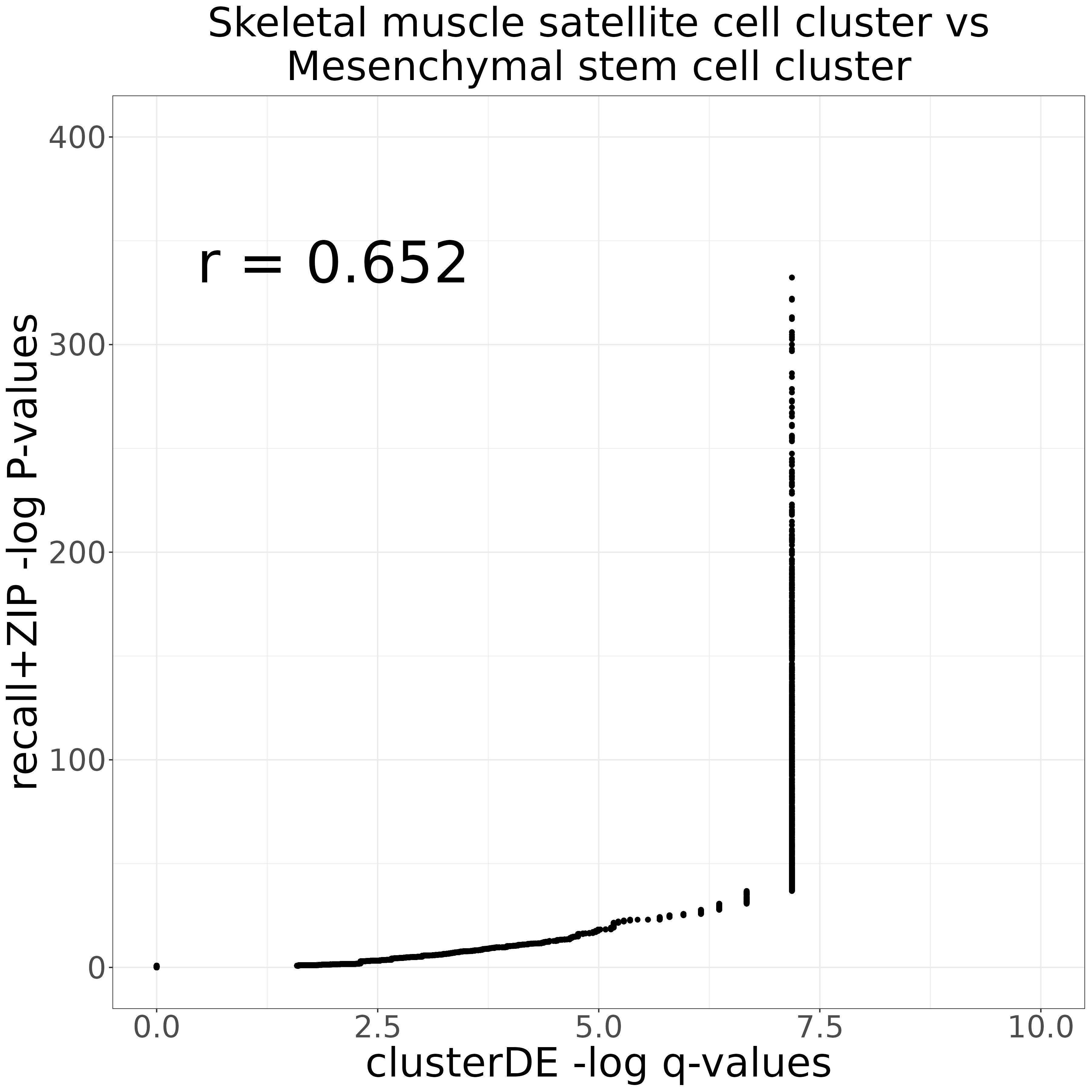
clusterDE_vs_recall_markers_plot <- compare_markers_q_vals(clusterDE_res$summaryTable, recall_markers_0vs1,
"clusterDE -log q-values",
"recall+ZIP -log P-values",
"Skeletal muscle satellite cell cluster vs\nMesenchymal stem cell cluster",
xmax = 10,
ymax = 400)
ggsave("countsplit_vs_recall_markers_plot.png", countsplit_vs_recall_markers_plot, width = 12, height = 12, units = "in")
ggsave("clusterDE_vs_recall_markers_plot.png", clusterDE_vs_recall_markers_plot, width = 12, height = 12, units = "in")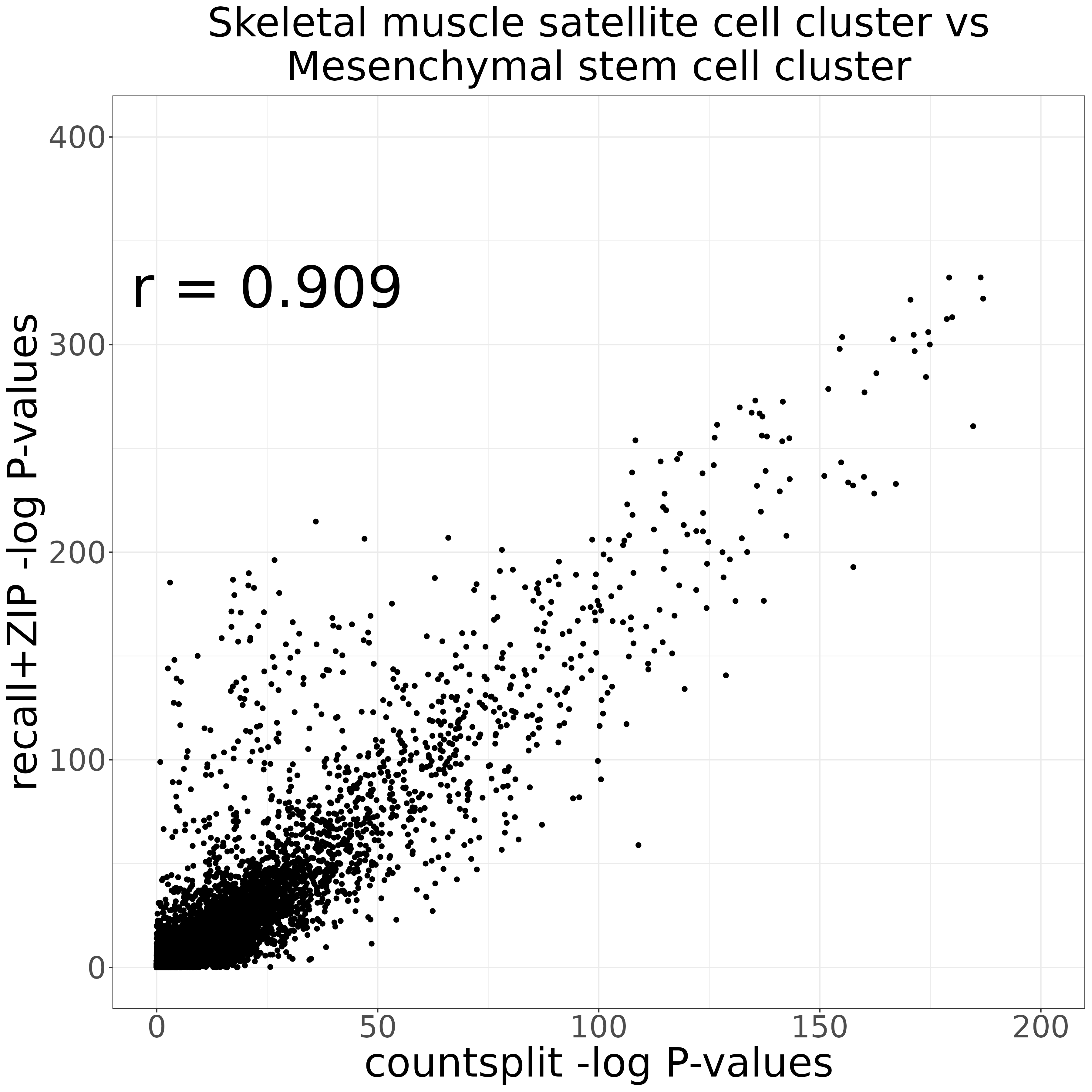
Plot the Venn diagram
countsplit_signficant <- countsplit_markers_0vs1[countsplit_markers_0vs1$p_val_adj < 0.05, ]
recall_signficant <- recall_markers_0vs1[recall_markers_0vs1$p_val_adj < 0.05, ]
marker_venn_diagram <- marker_gene_venn_diagram(rownames(recall_signficant), rownames(countsplit_signficant), clusterDE_res$DEgenes)
ggsave("marker_venn_diagram.png", marker_venn_diagram)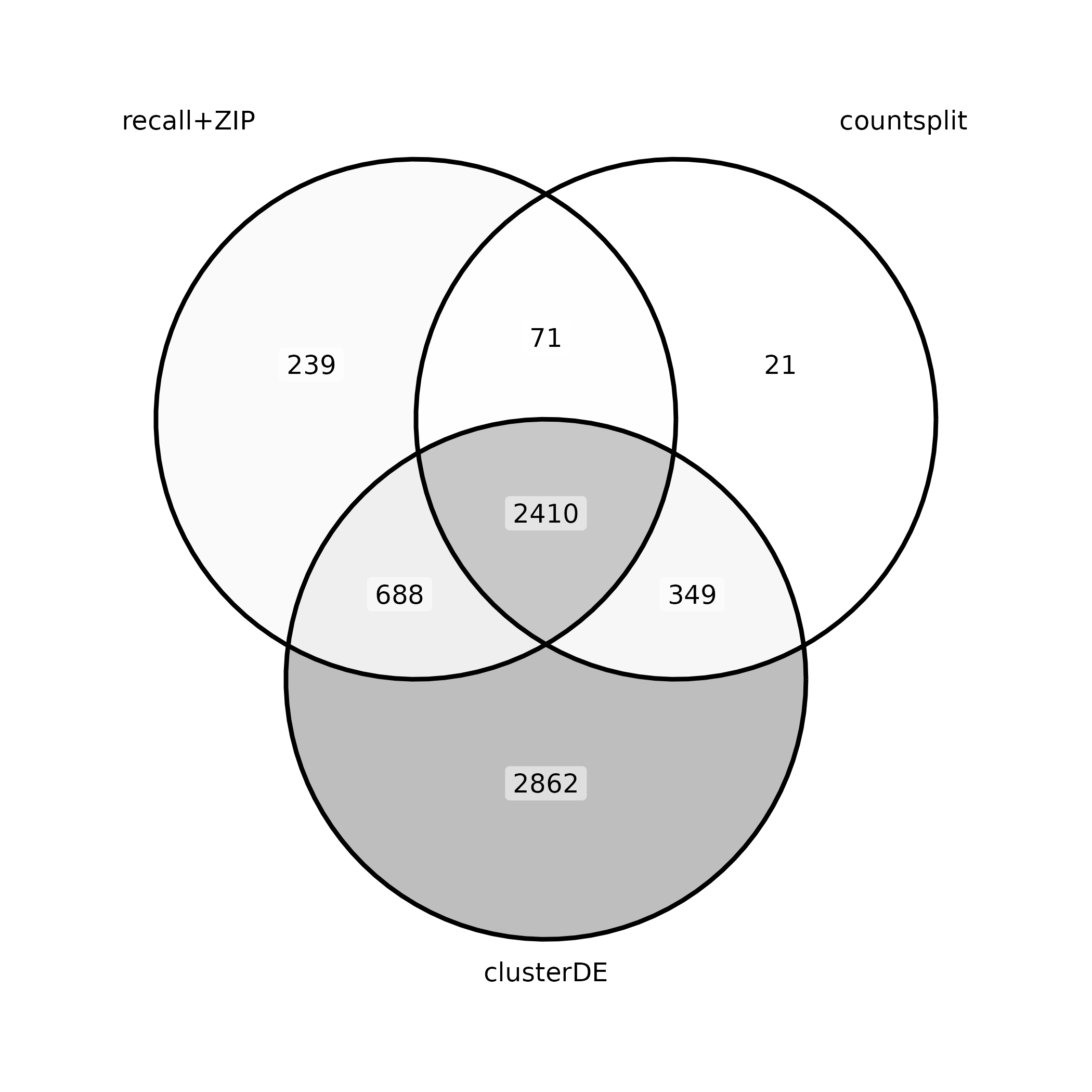
Finally, we plot the P-value comparison for recall and countsplit with recall clusters downsampled.
Idents(limb_muscle) <- limb_muscle@meta.data$recall_idents
cluster_0_downsampled_indices <- WhichCells(limb_muscle, idents = c("0"), downsample = 271)
cluster_1_downsampled_indices <- WhichCells(limb_muscle, idents = c("1"), downsample = 192)
recall_downsampled_indices <- c(cluster_0_downsampled_indices, cluster_1_downsampled_indices)
limb_muscle_downsampled <- limb_muscle[, recall_downsampled_indices]
recall_downsampled_markers <- FindMarkers(limb_muscle_downsampled,ident.1 = 0, ident.2 = 1, min.pct = 0, logfc.threshold = 0)
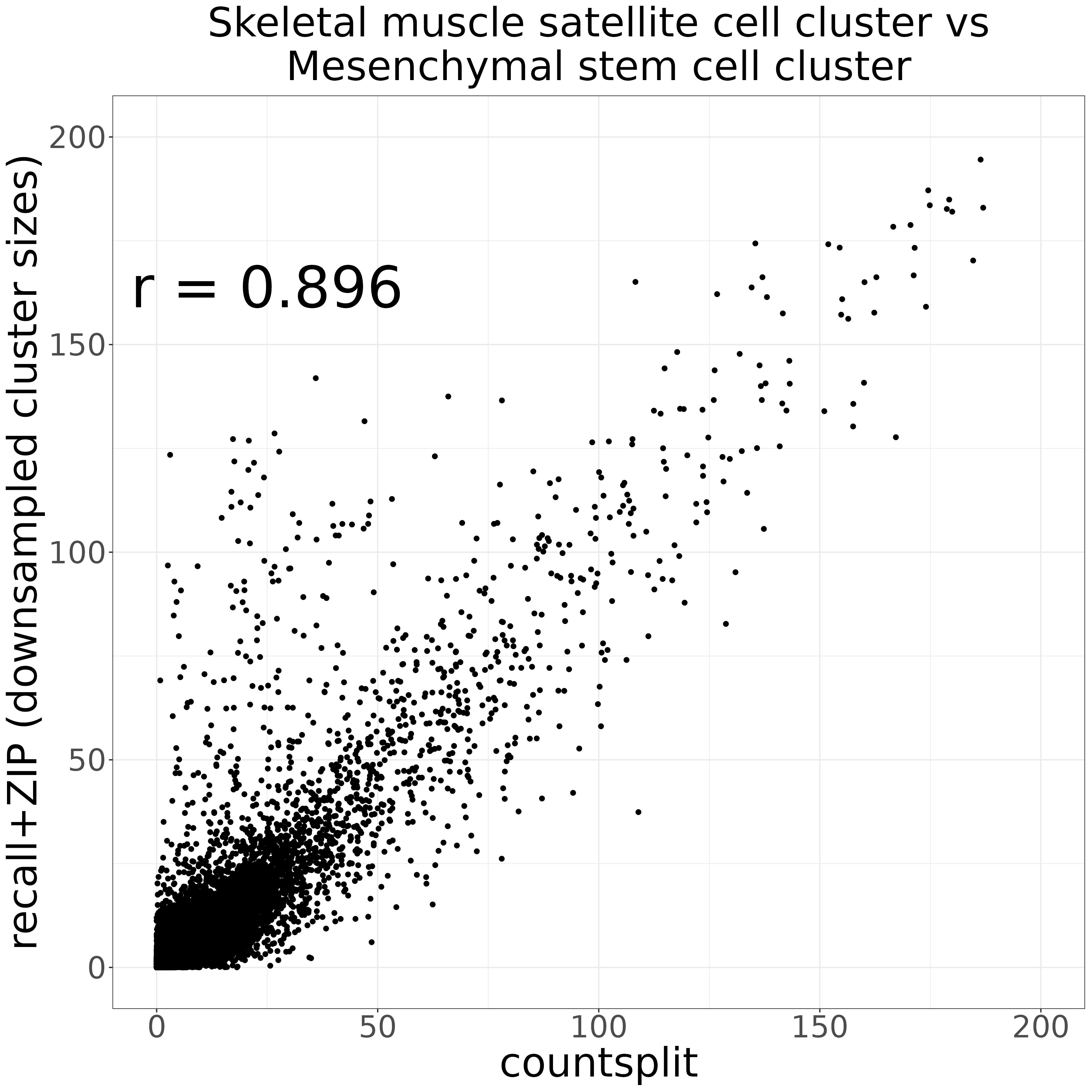
recall_downsampled_vs_counsplit_markers_plot <- compare_markers_p_vals(countsplit_markers_0vs1,
recall_downsampled_markers,
xlabel = "countsplit",
ylabel = "recall+ZIP (downsampled cluster sizes)",
title = "Skeletal muscle satellite cell cluster vs\nMesenchymal stem cell cluster",
xmax = 200,
ymax = 200)
ggsave("recall_downsampled_vs_counsplit_markers_plot.png", recall_downsampled_vs_counsplit_markers_plot, width = 12, height = 12, units = "in")