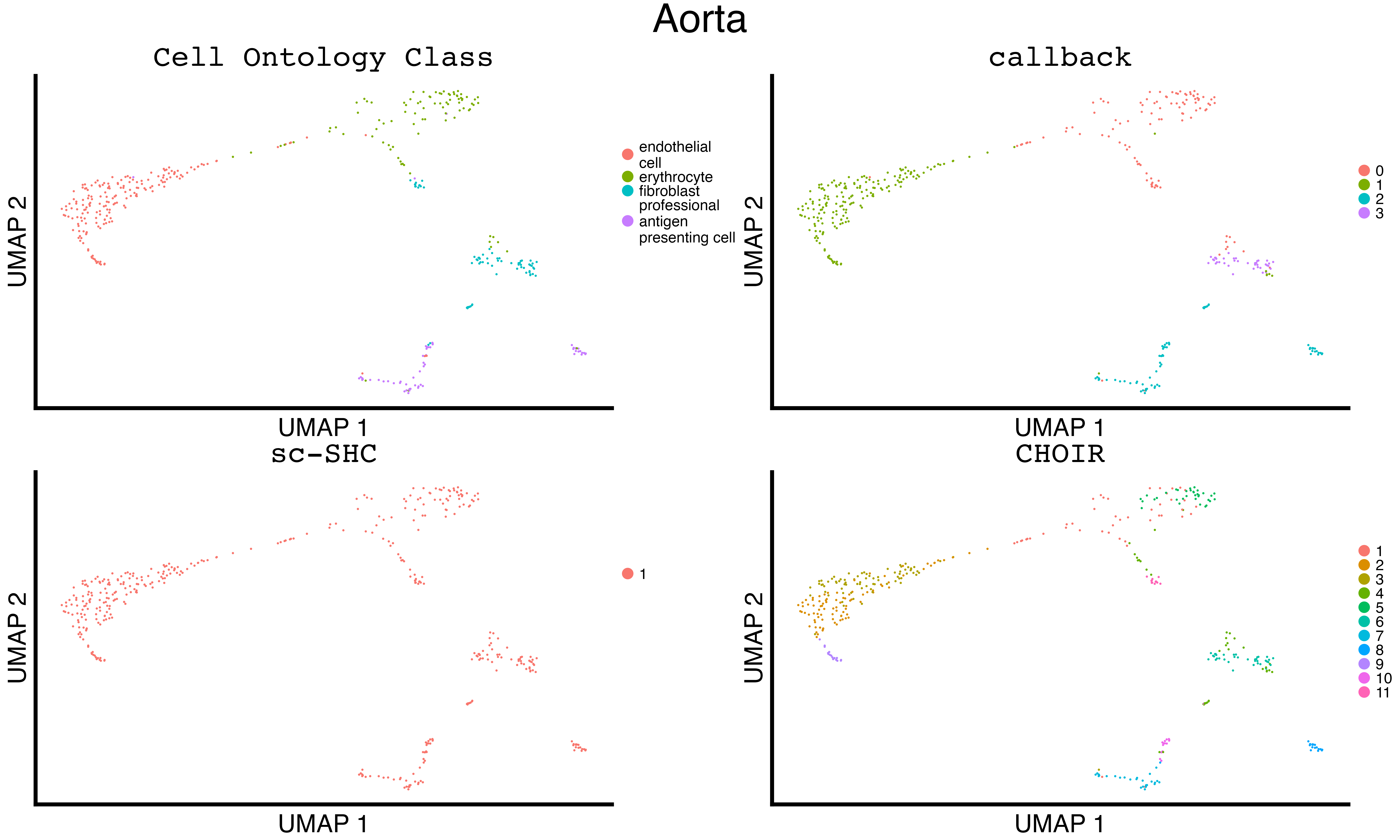
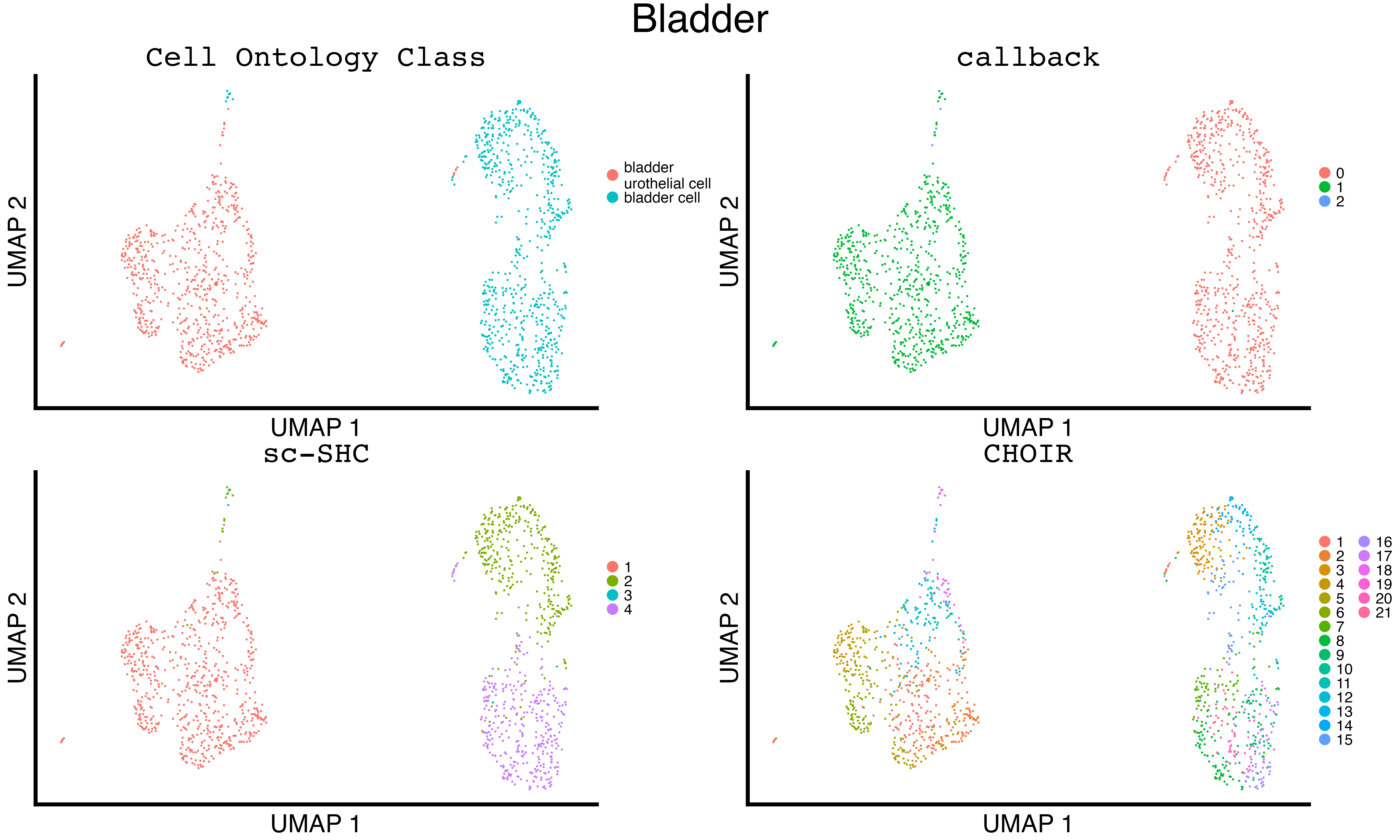
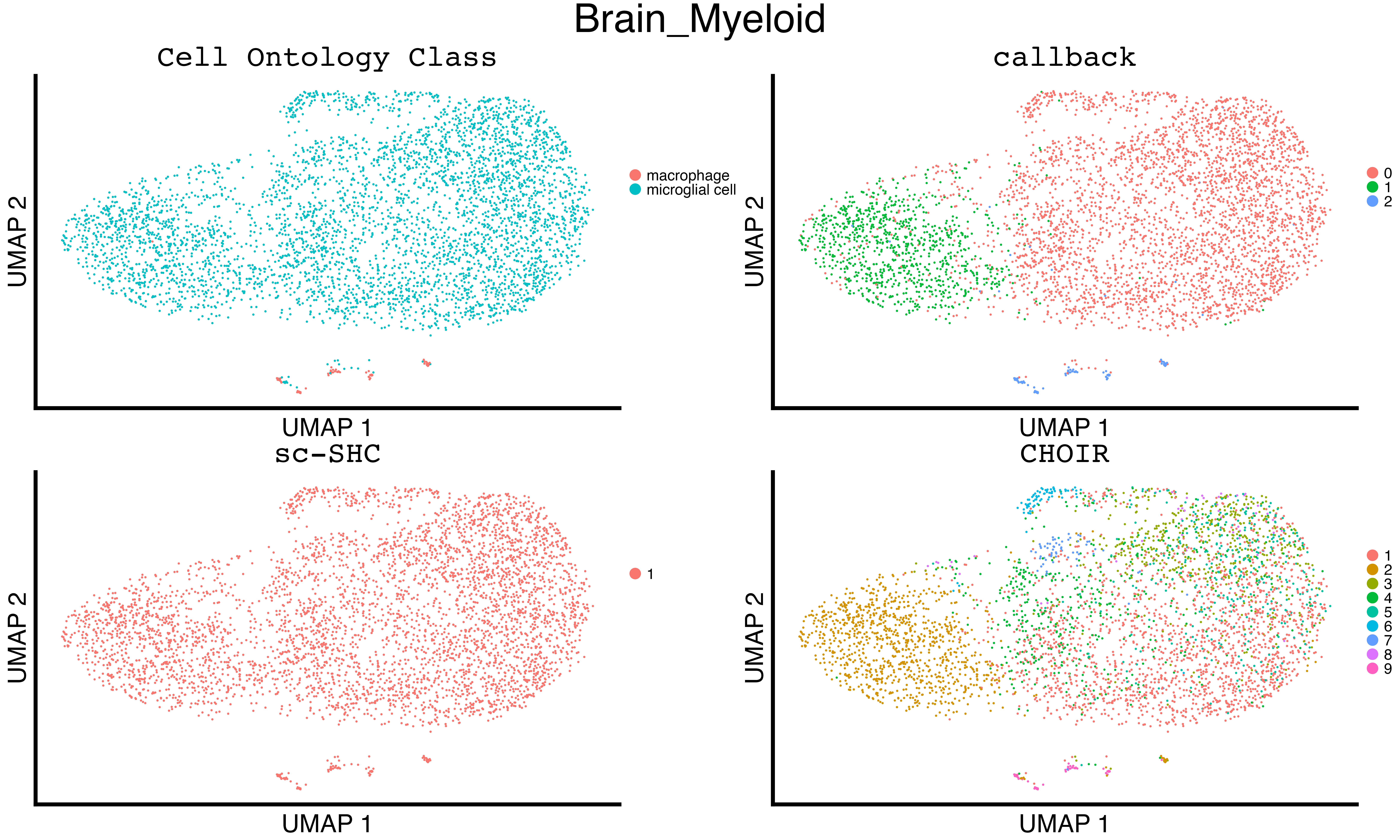
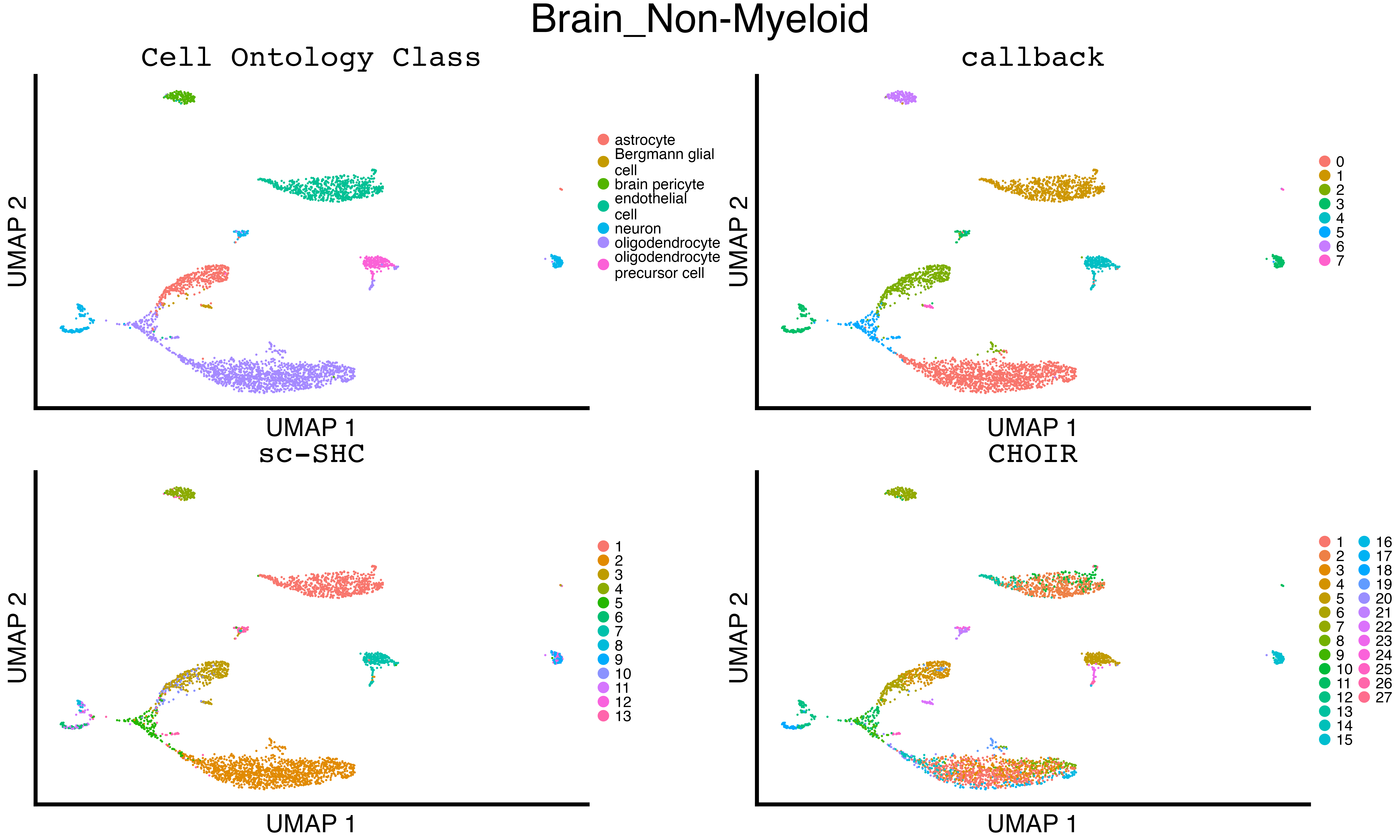
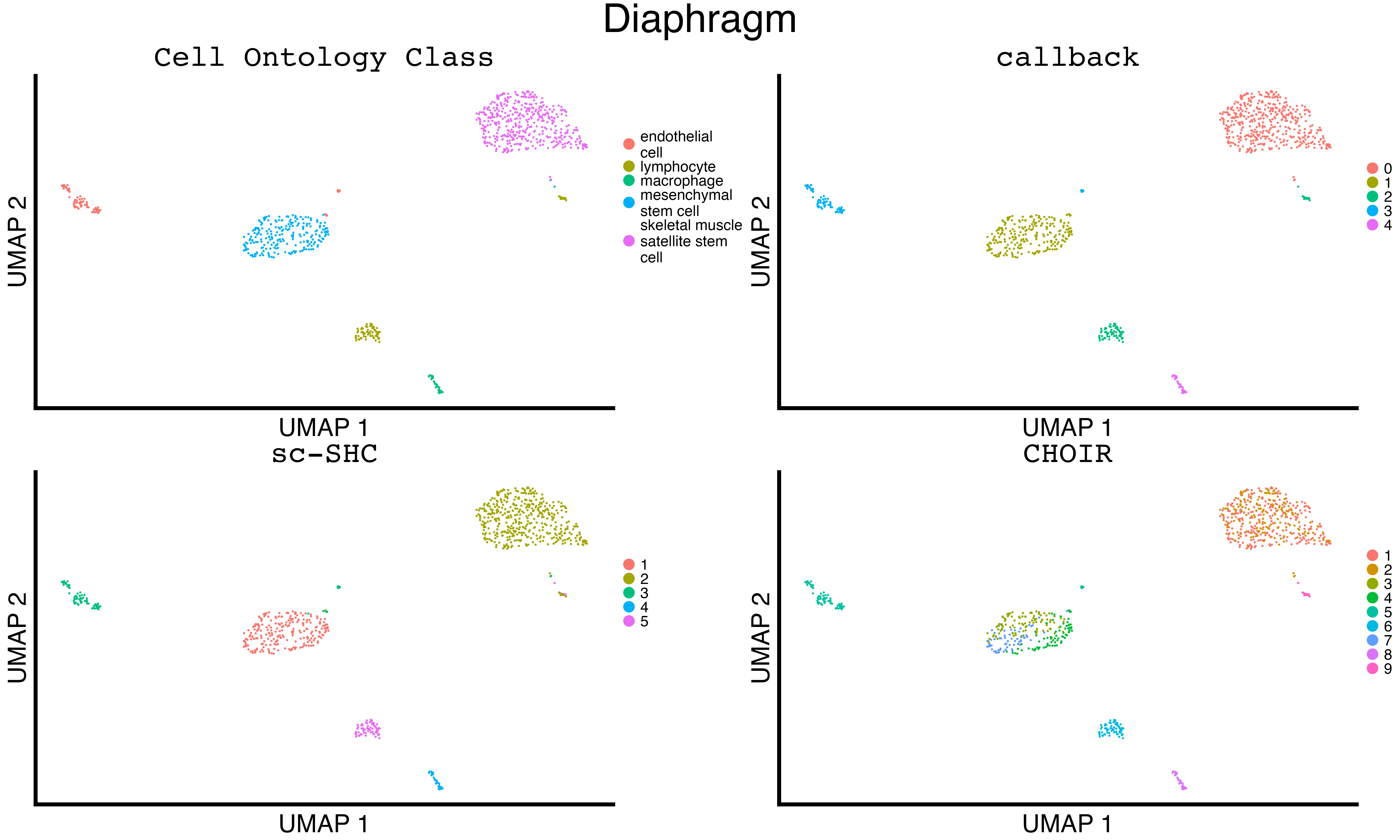
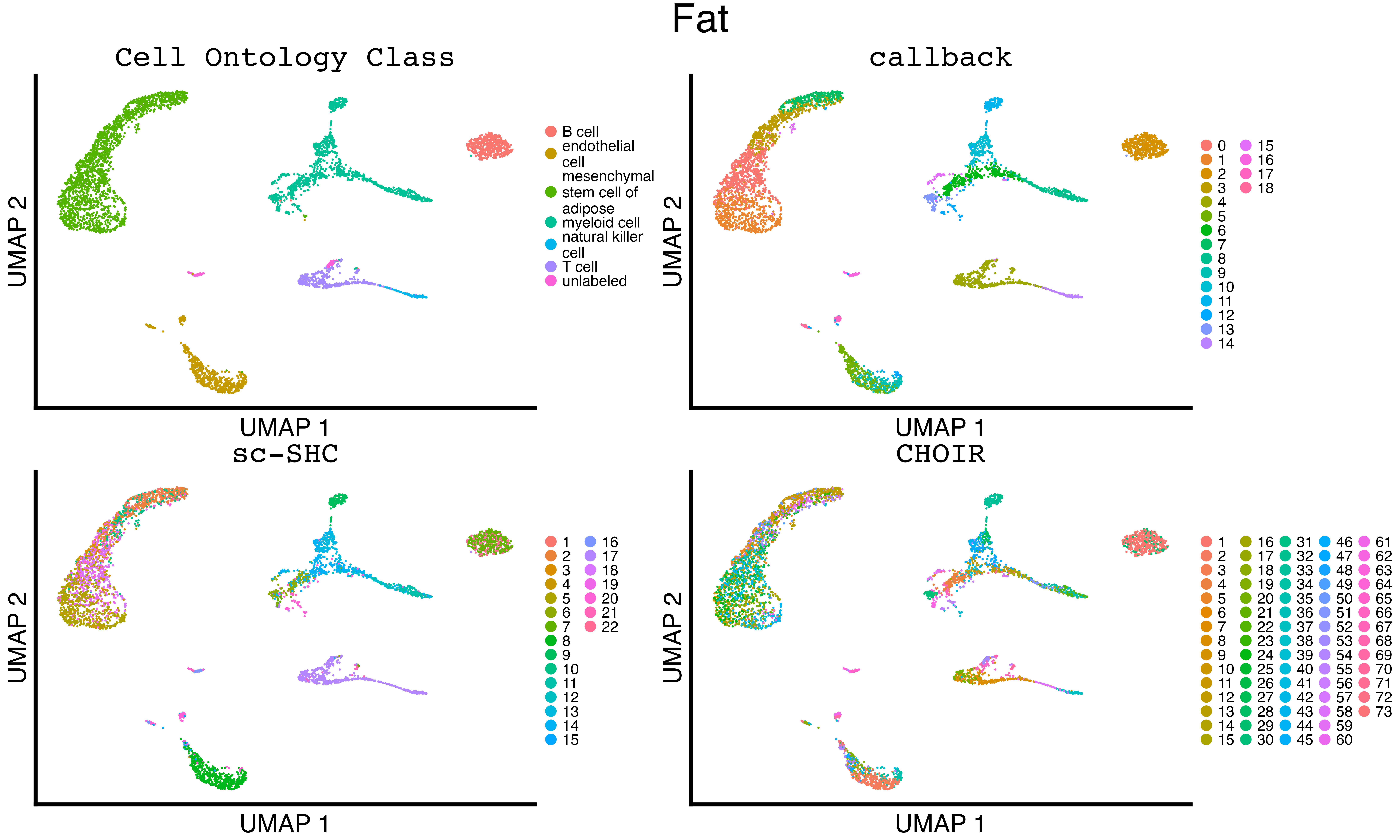
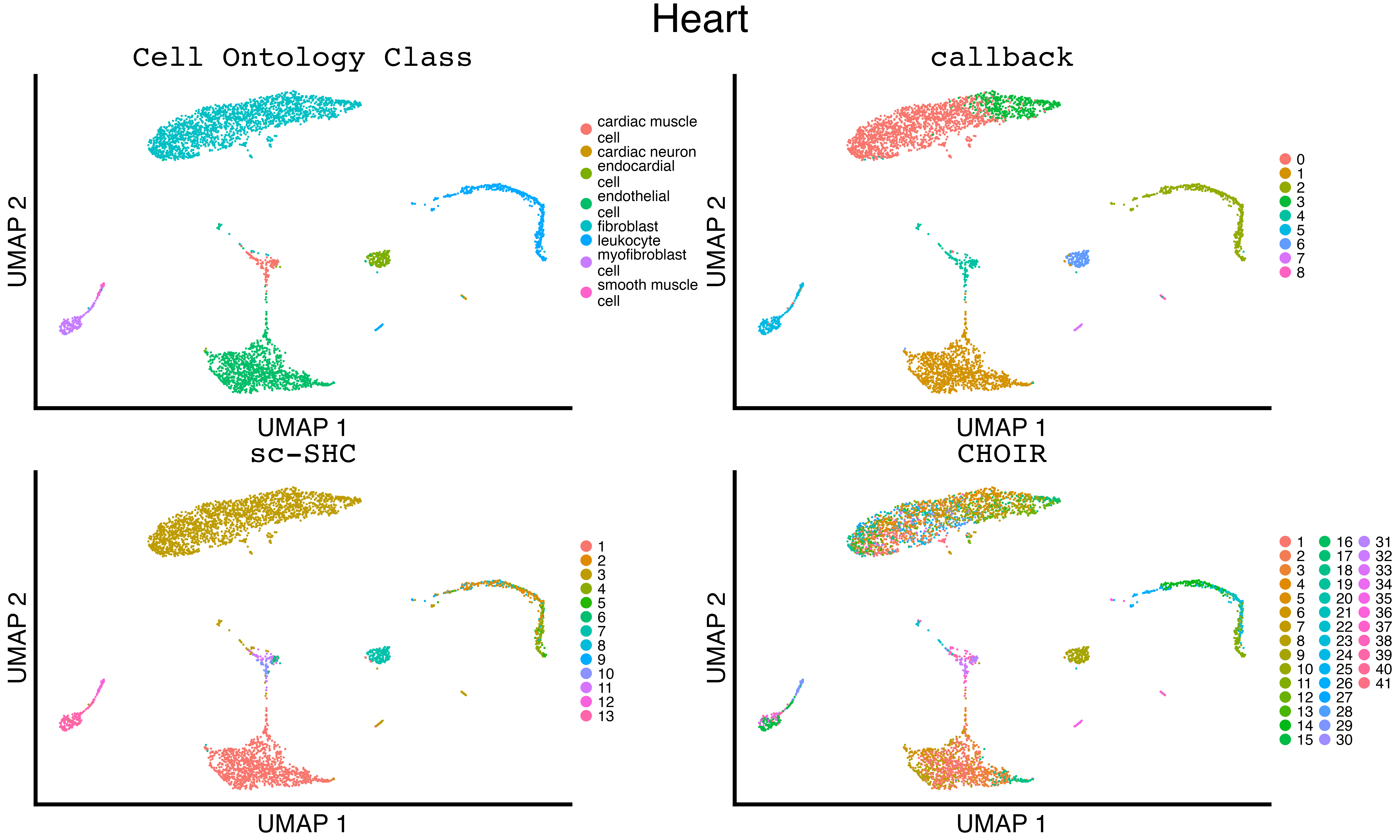
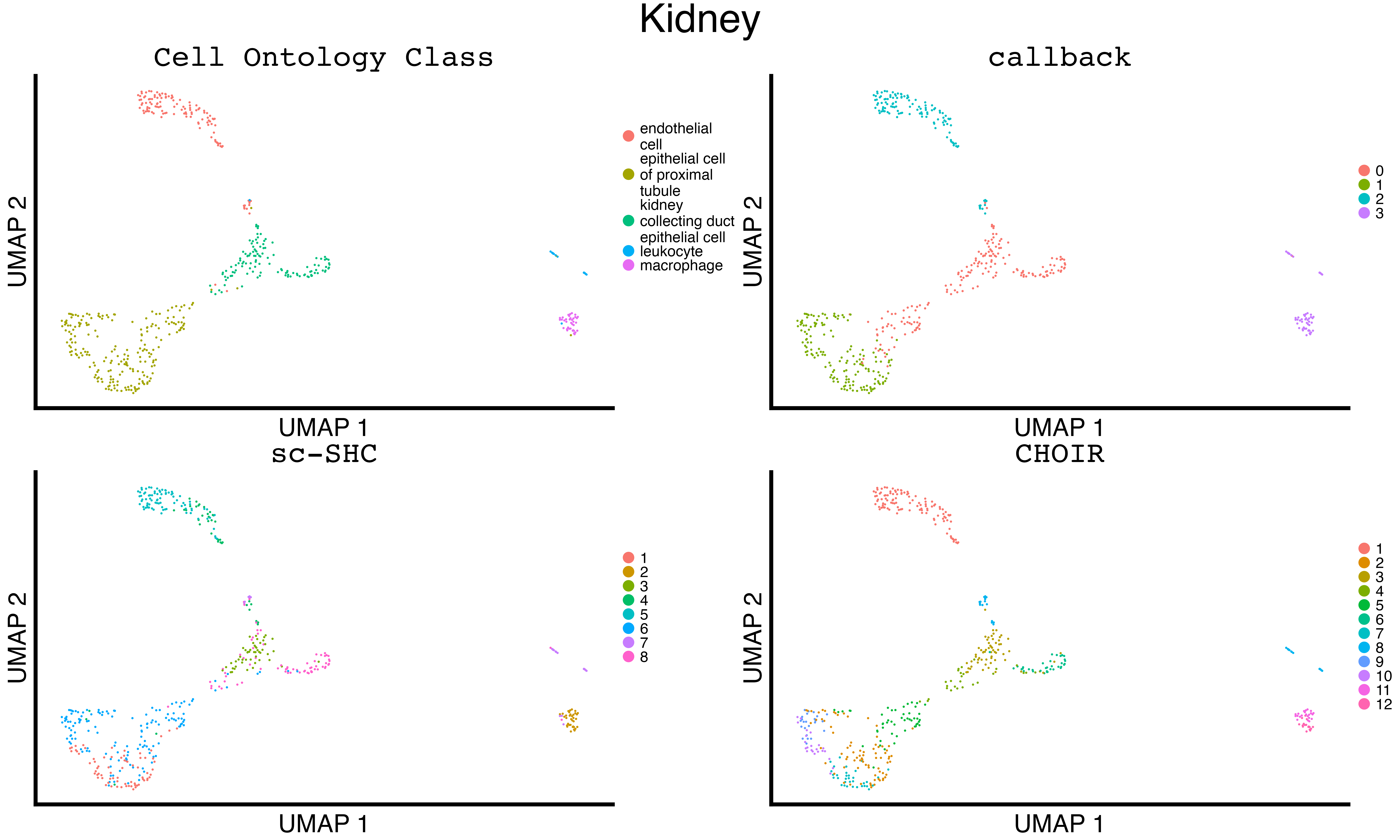
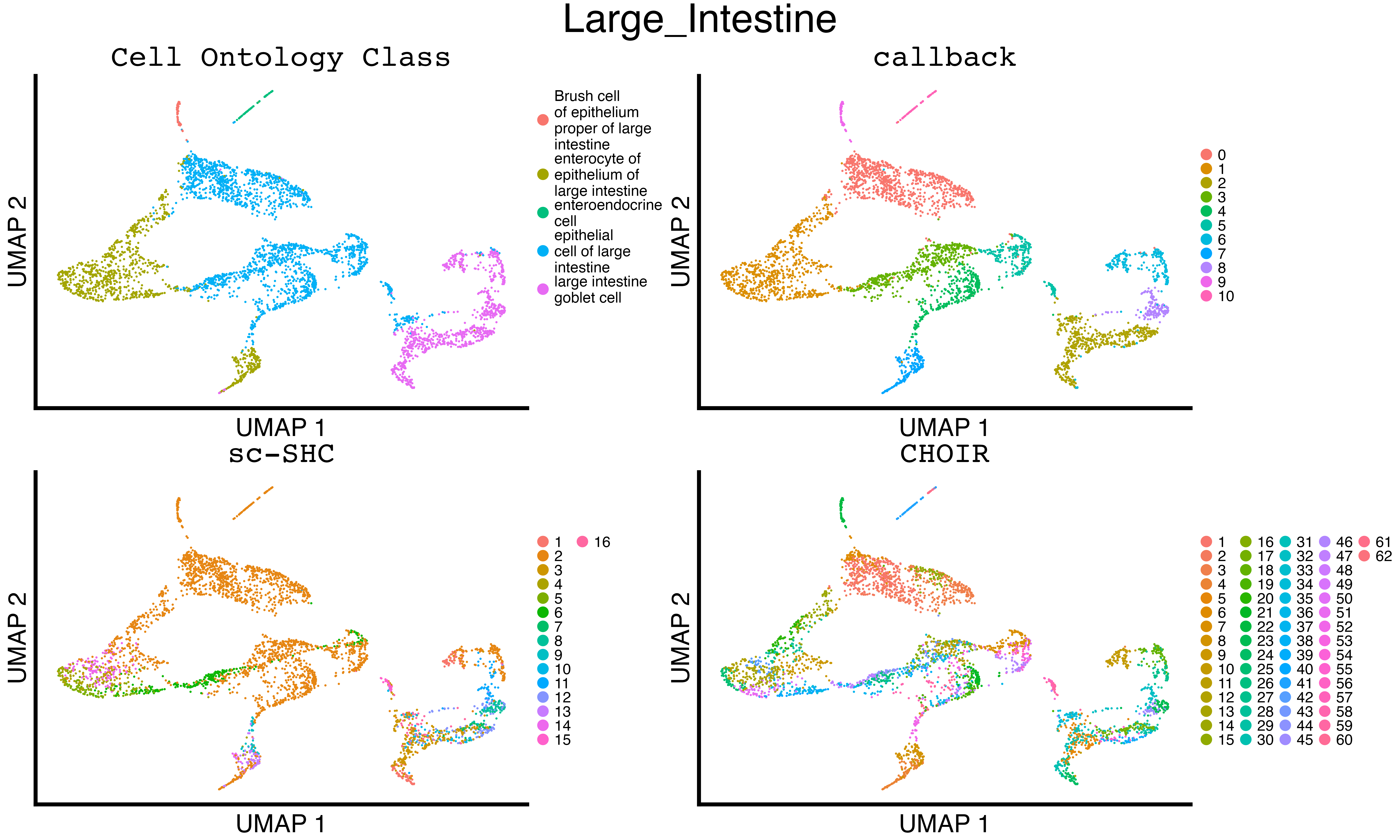
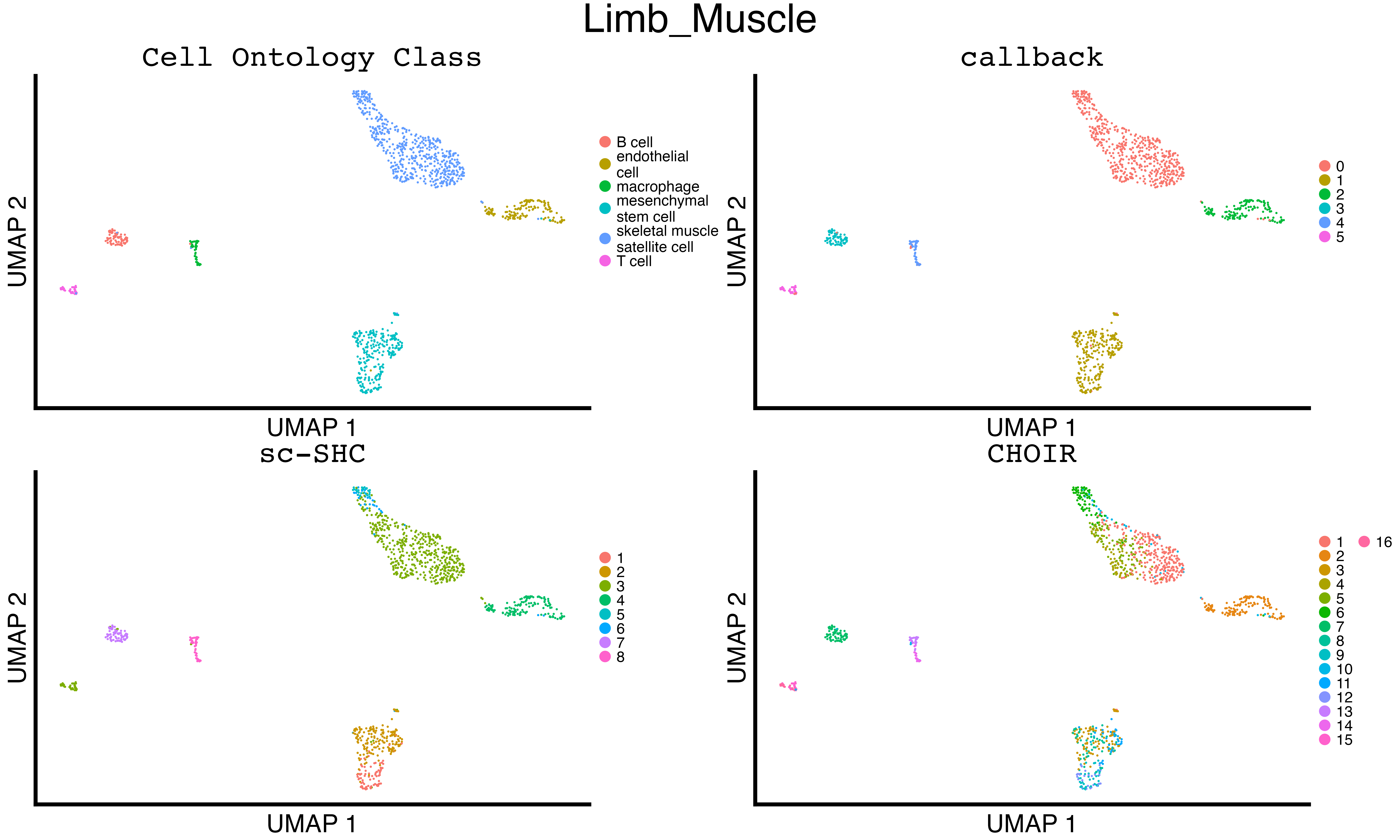
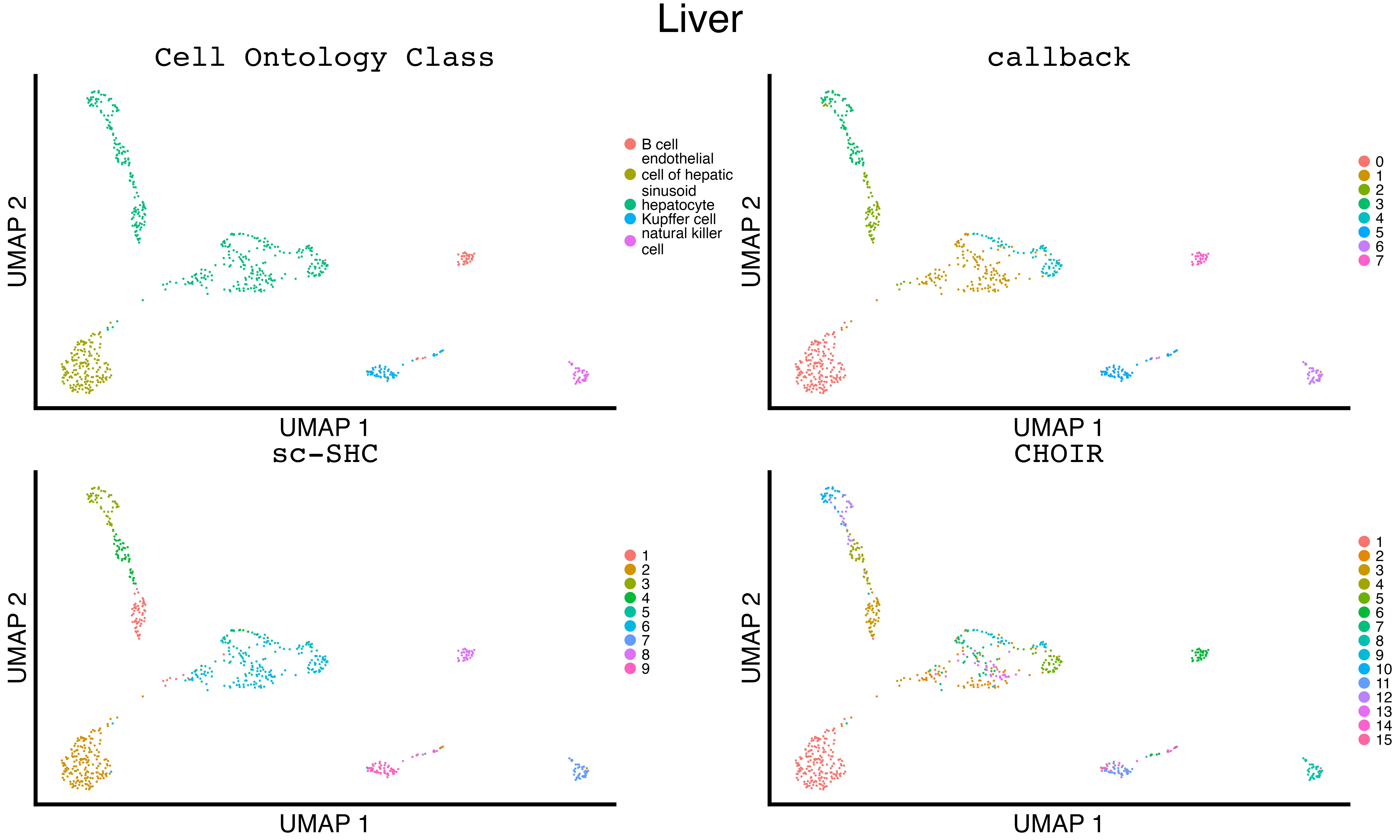
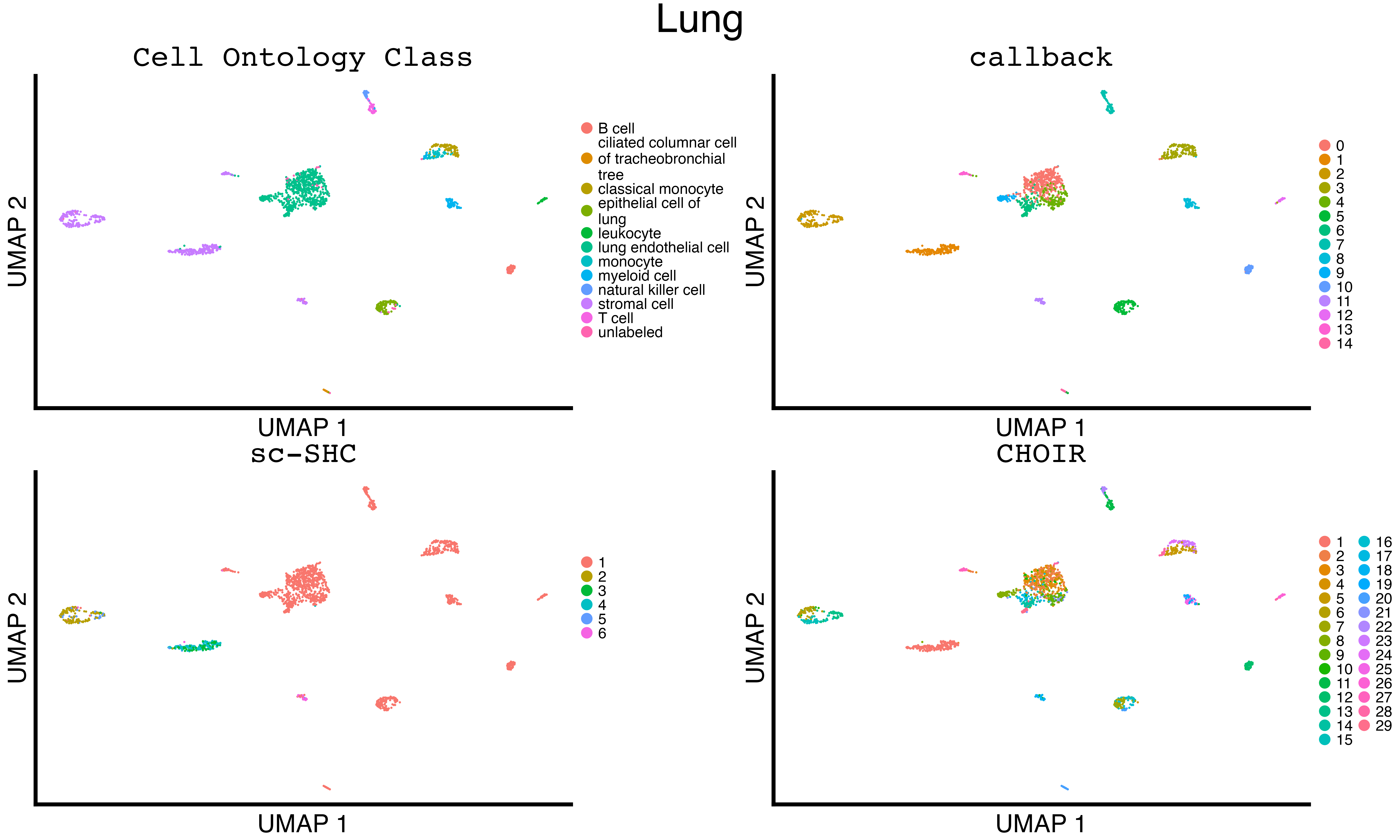
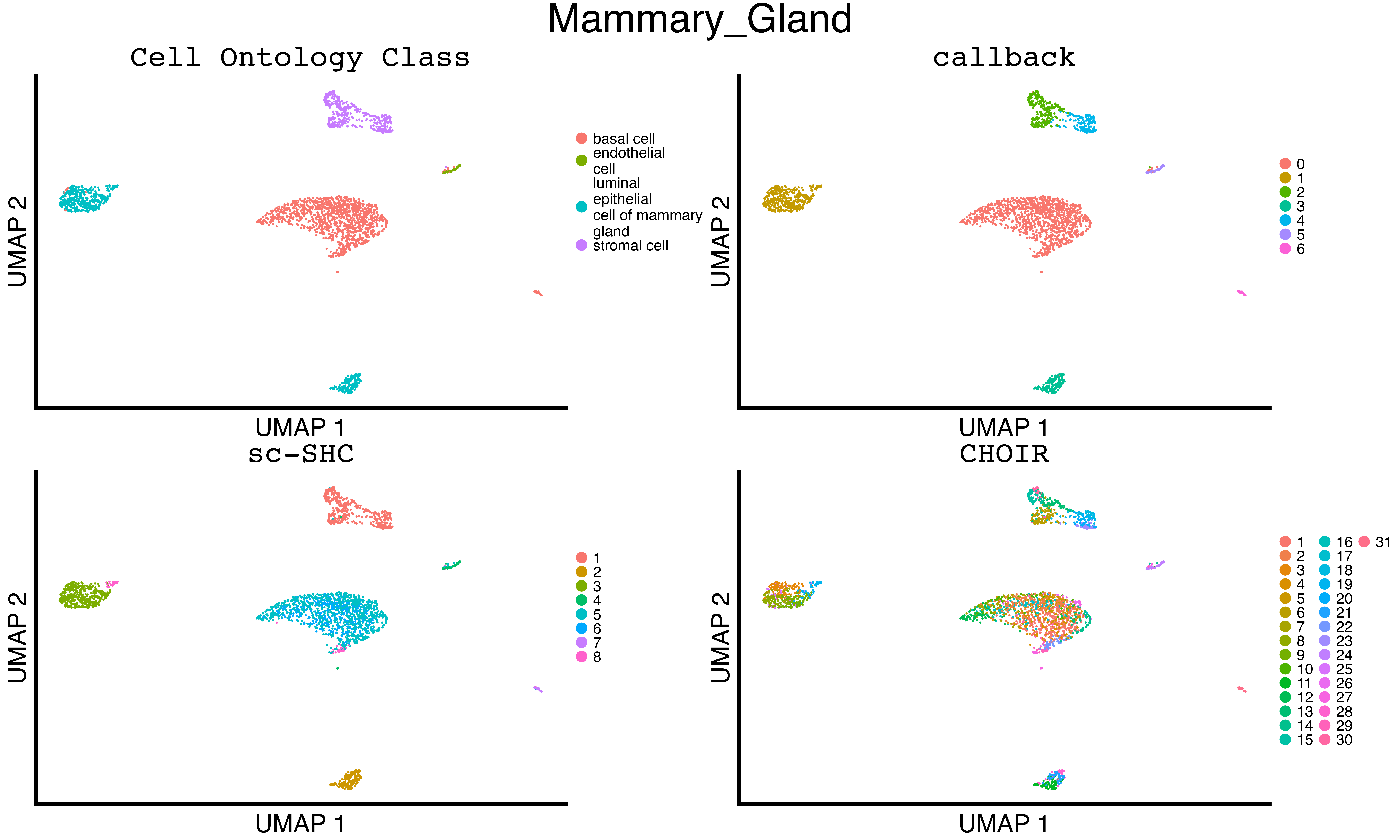
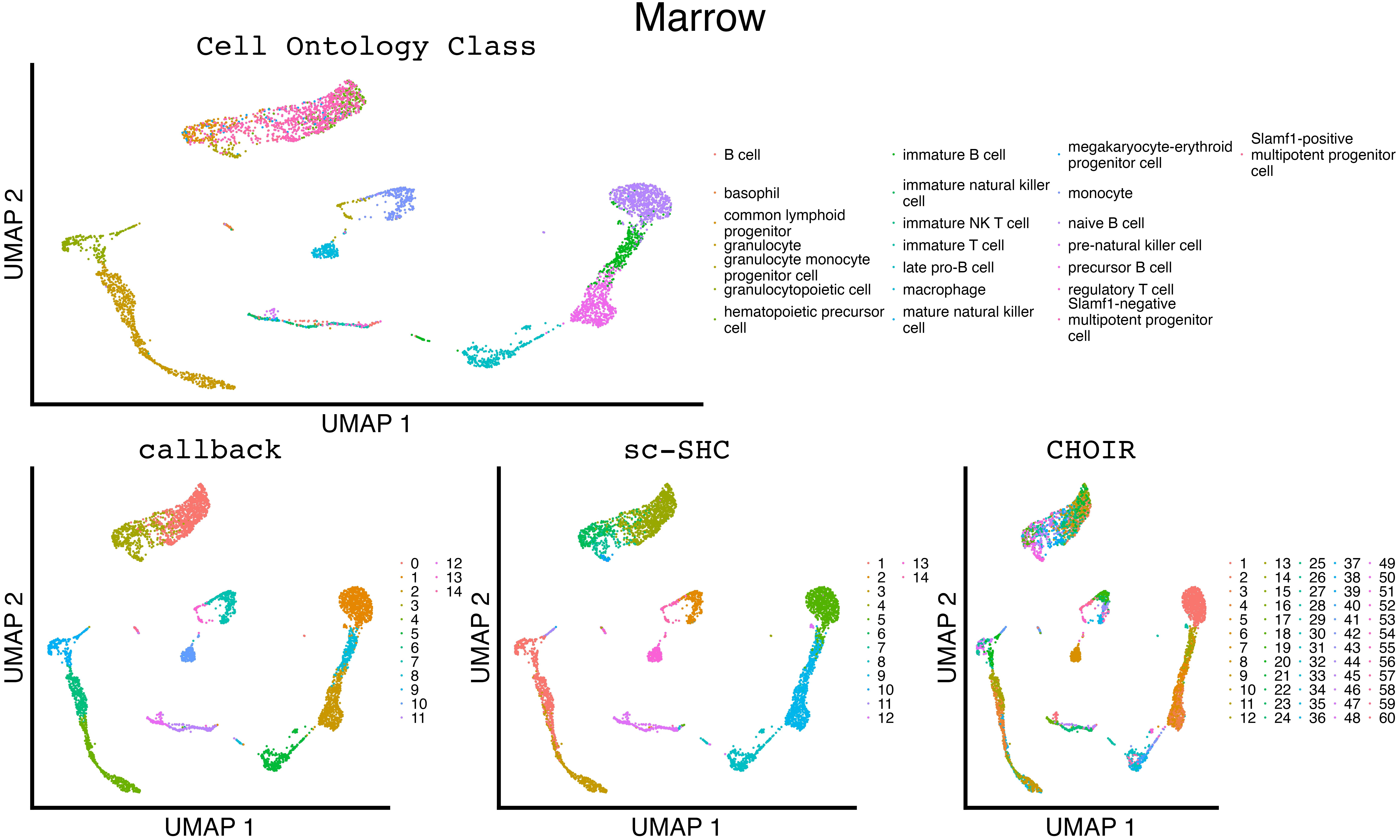
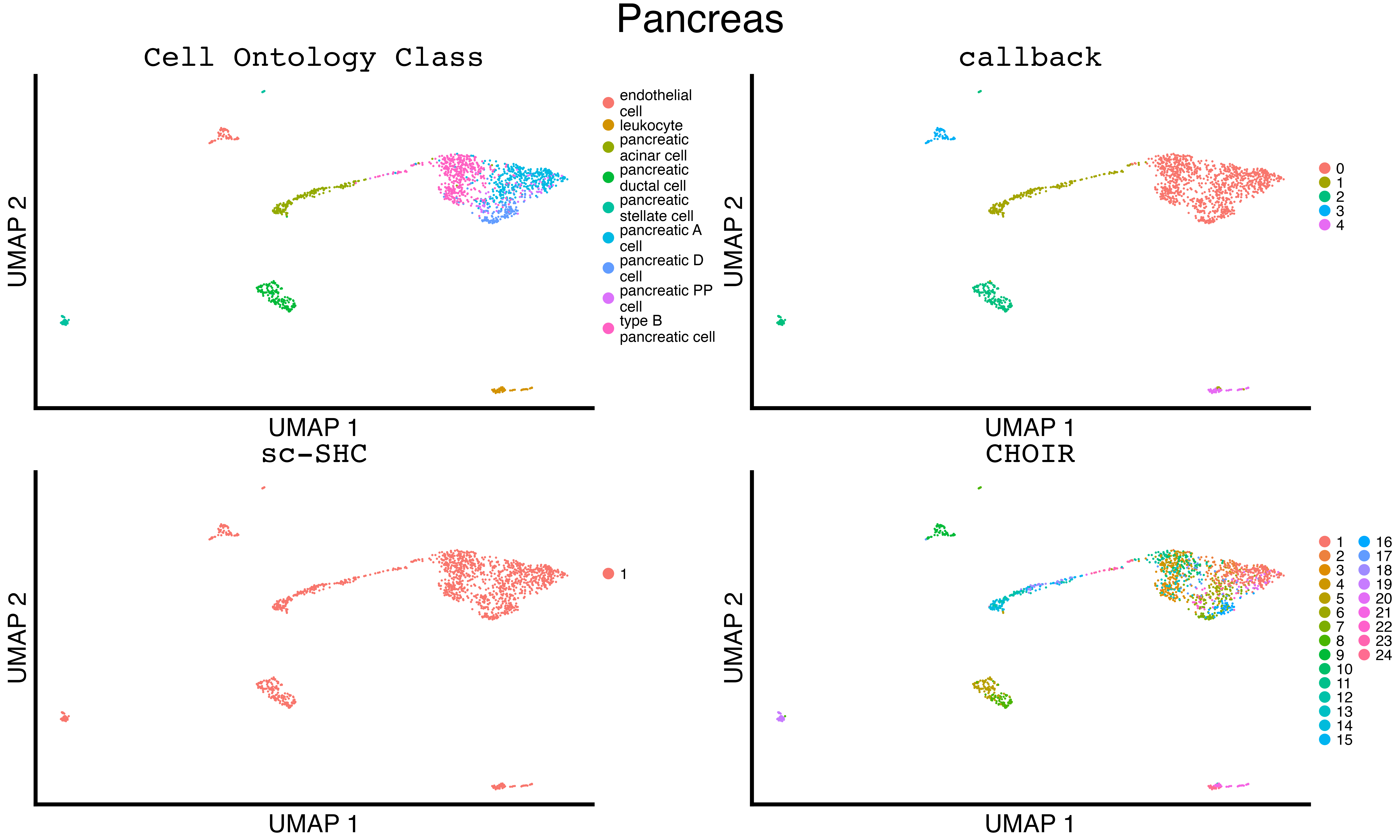
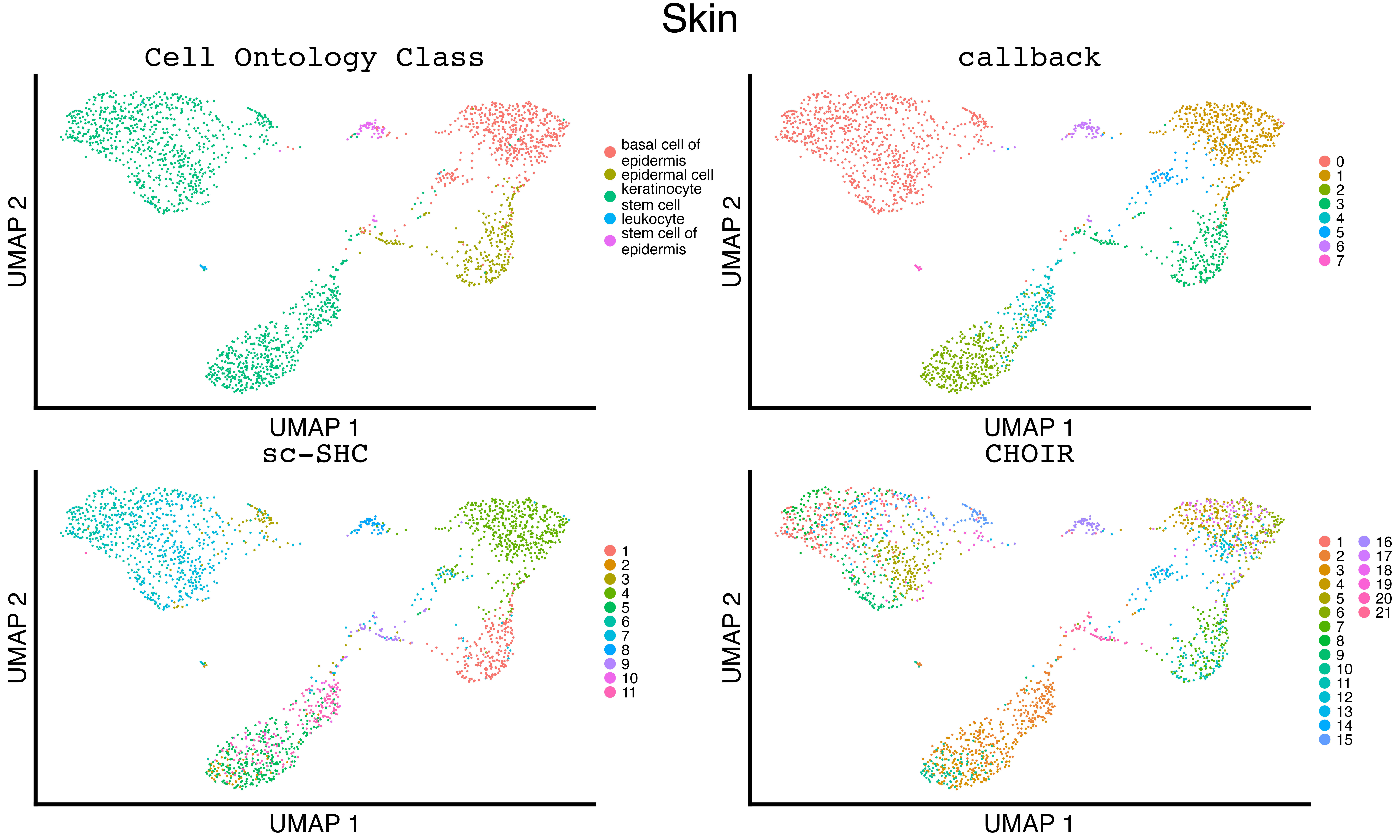
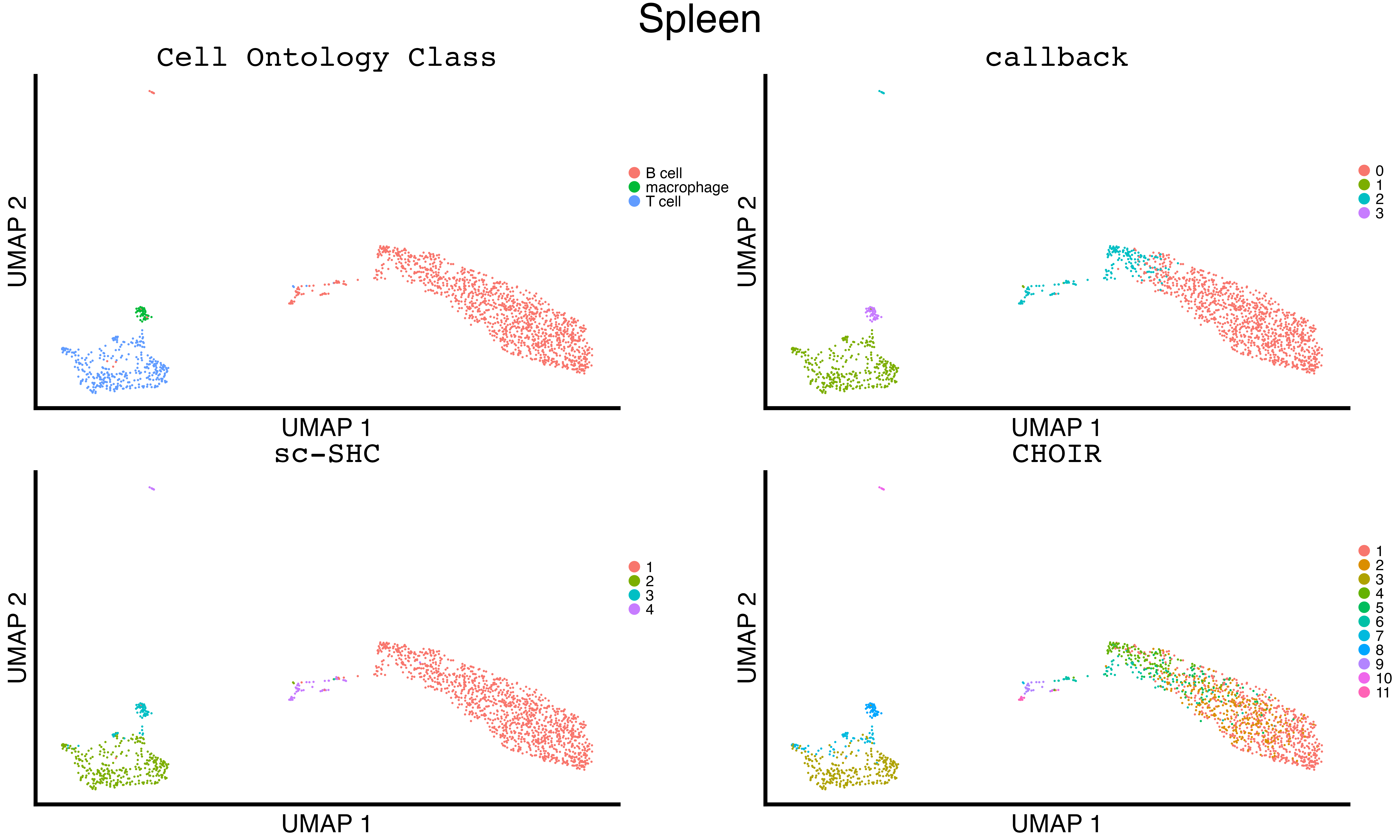
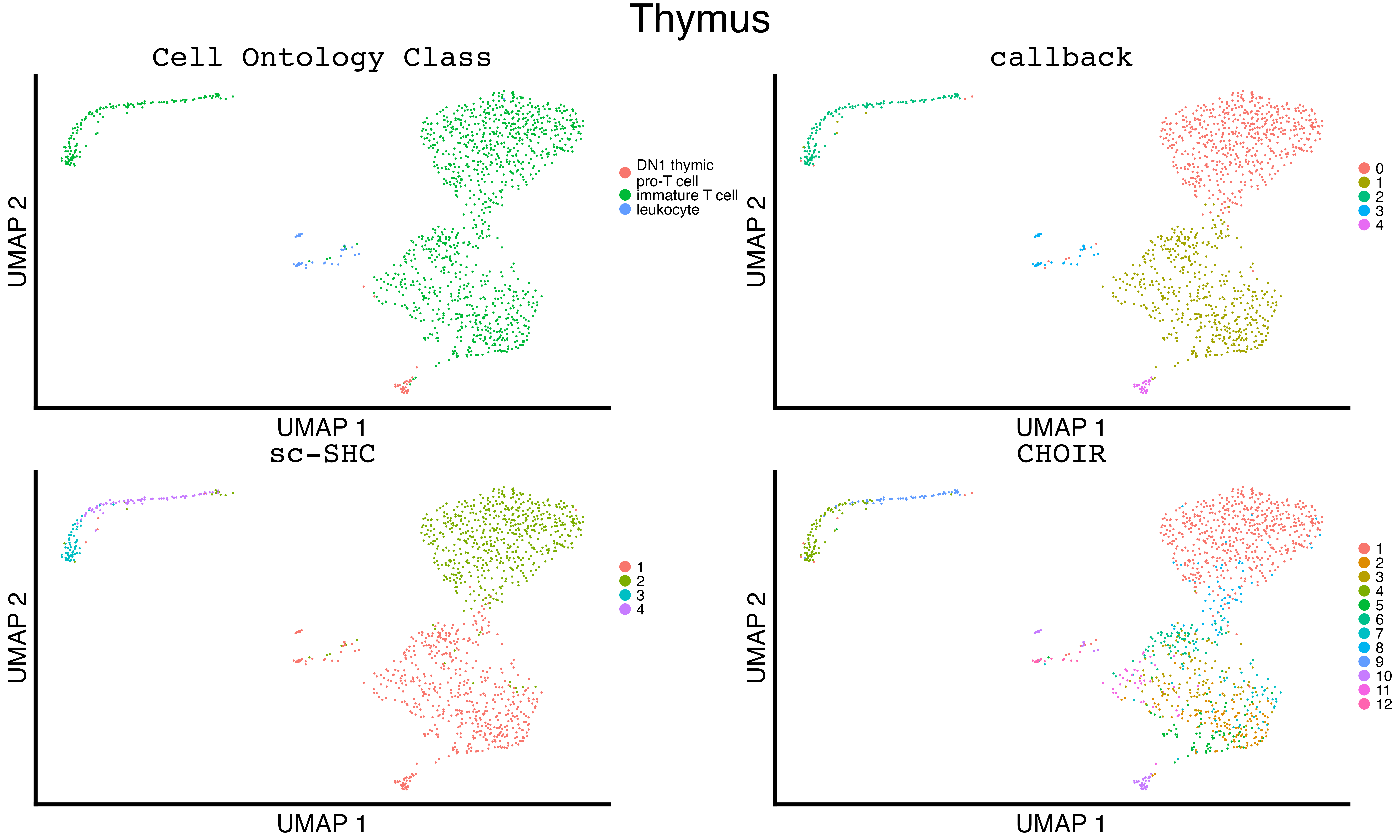
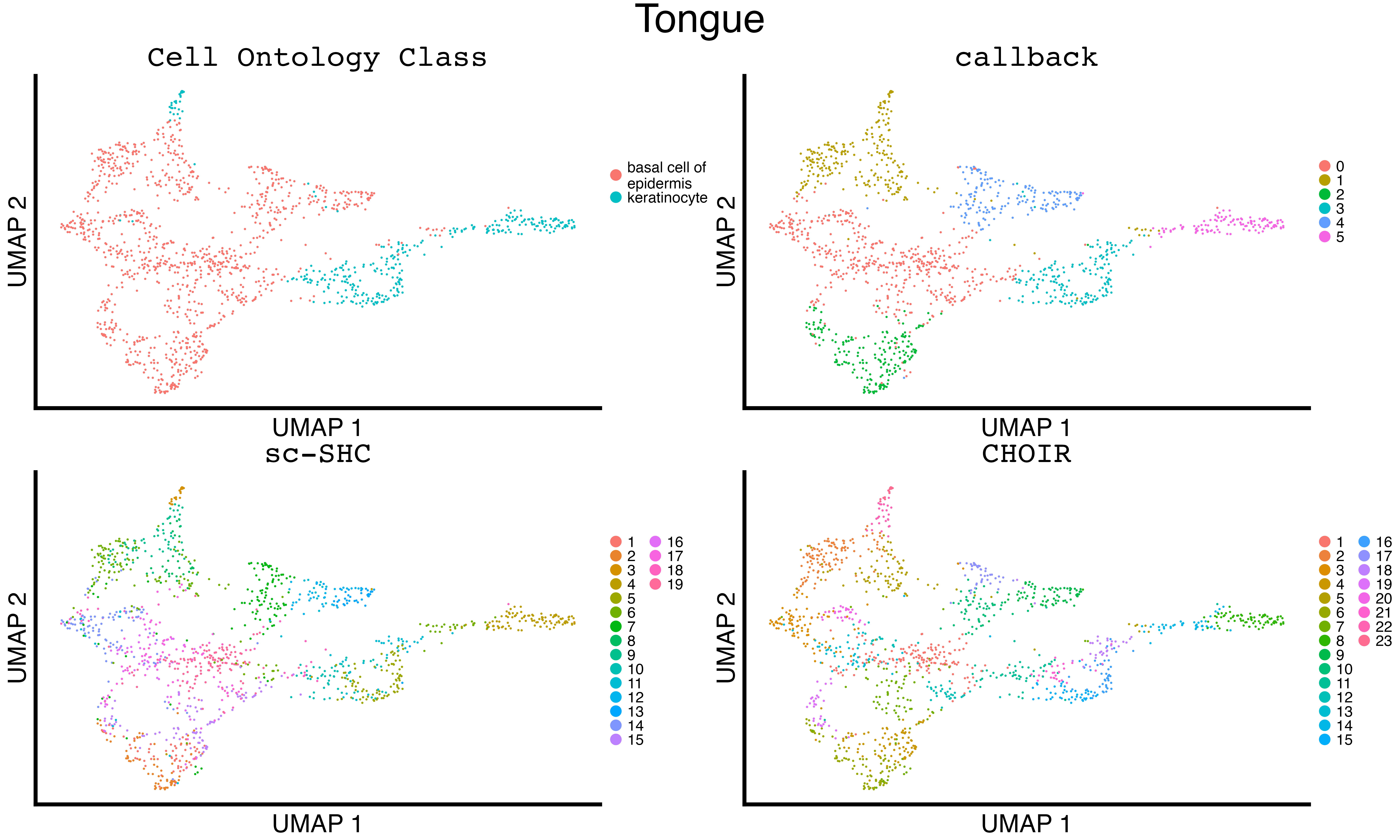
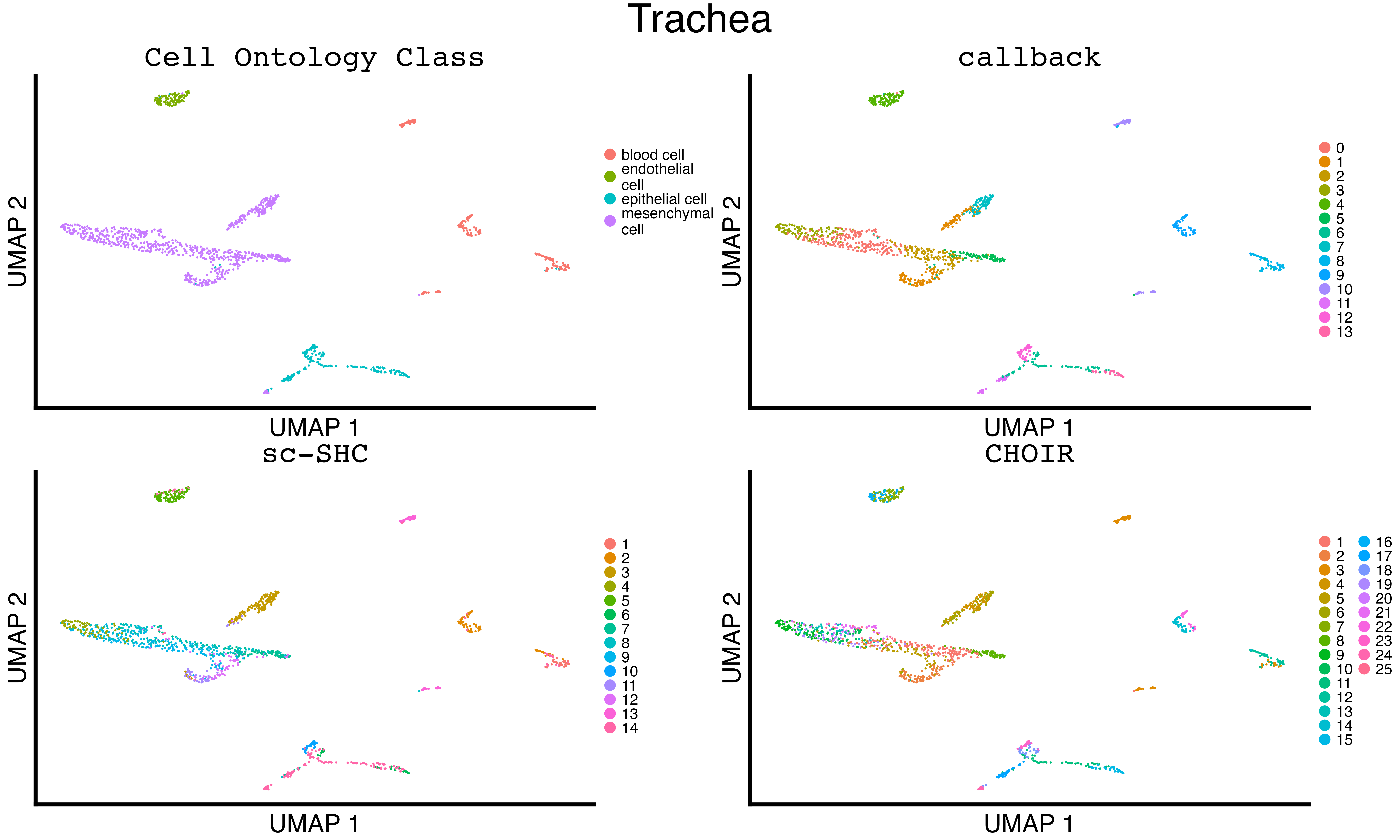
6. Plotting Tabula Muris UMAPs (Supplemental Figures S6-S25)
6_tissue_umaps.Rmd
suppressPackageStartupMessages({
library(callbackreproducibility)
library(Seurat)
library(ggplot2)
library(patchwork)
library(grid)
library(gridExtra)
})Get the clustering results from running callback,
sc-SHC, and CHOIR.
tissue_seurat_files <- list.files(pattern = "cluster_results_seurat.rds")Loop over the clustering results and plot all of the UMAP grids.
for (tissue_rds_file in tissue_seurat_files) {
tissue_name <- sub('cluster_results_seurat.rds', "", tissue_rds_file)
print(tissue_name)
tissue_seurat_obj <- readRDS(tissue_rds_file)
if (tissue_name == "Heart") {
# clean up cell type labels for heart
tissue_seurat_obj@meta.data$cell_ontology_class[tissue_seurat_obj@meta.data$cell_ontology_class == ""] <- "cardiac neuron"
}
if (tissue_name == "Fat") {
# clean up cell type labels for fat
tissue_seurat_obj@meta.data$cell_ontology_class[tissue_seurat_obj@meta.data$cell_ontology_class == ""] <- "unlabeled"
}
if (tissue_name == "Lung") {
# clean up cell type labels for heart
tissue_seurat_obj@meta.data$cell_ontology_class[tissue_seurat_obj@meta.data$cell_ontology_class == ""] <- "unlabeled"
}
# make legends fit better
legend_line_length <- 15
if (tissue_name == "Lung") {
legend_line_length <- 22
}
# make some cell types multiple lines
tissue_seurat_obj@meta.data$cell_ontology_class <- stringr::str_wrap(tissue_seurat_obj@meta.data$cell_ontology_class, legend_line_length)
# remove NAs
tissue_seurat_obj <- subset(tissue_seurat_obj, subset = cell_ontology_class %in% levels(factor(tissue_seurat_obj@meta.data$cell_ontology_class)))
umap_fig <- get_umap(tissue_seurat_obj, tissue_name)
if (tissue_name == "Marrow") {
legend_line_length <- 23
tissue_seurat_obj@meta.data$cell_ontology_class <- stringr::str_wrap(tissue_seurat_obj@meta.data$cell_ontology_class, legend_line_length)
layout <- "AAA###\nAAA###\nBBCCDD\nBBCCDD"
marrow_ontology <- one_umap(tissue_seurat_obj, "cell_ontology_class", "Cell Ontology Class", add_legend=TRUE) + guides(color=guide_legend(nrow=7))
marrow_callback <- one_umap(tissue_seurat_obj, "callback_idents", "callback", add_legend=TRUE) + guides(color=guide_legend(nrow=12))
marrow_scSHC <- one_umap(tissue_seurat_obj, "scSHC_clusters", "sc-SHC", add_legend=TRUE) + guides(color=guide_legend(nrow=12))
marrow_CHOIR <- one_umap(tissue_seurat_obj, "CHOIR_clusters_0.05", "CHOIR", add_legend=TRUE) + guides(color=guide_legend(nrow=12))
umap_fig <- marrow_ontology +
marrow_callback +
marrow_scSHC +
marrow_CHOIR +
plot_layout(design = layout) +
plot_annotation(title = tissue_name,
theme = theme(plot.title = element_text(size = 64, hjust = 0.5)))
#umap_fig <- grid.arrange(
umap_fig <- arrangeGrob(
marrow_ontology, marrow_callback, marrow_scSHC, marrow_CHOIR,
# widths = c(2, 1, 1),
top=textGrob("Marrow", gp=gpar(fontsize = 64, hjust = 0.5, vjust = 1.0)),
layout_matrix = rbind(c(1, 1, 1),
c(2, 3, 4))
)
}
ggsave(paste0(tissue_name, "_umap.png"), plot = umap_fig, height = 1.3 * 3 * 1440, width = 1.3 * 5 * 1440, units = "px")
}